... Dispasia Fibrosa ...
Discusión:
Displasia fibrosa es el término acuñado por
Lichtenstein y Jaffe para describir un trastorno que afecta fundamentalmente al
hueso, pero a veces se acompaña de anormalidades extraesqueléticas. El cuadro se
caracteriza por la presencia de tejidos fibroóseos en expansión, en el interior
de los huesos afectados. El
hueso normal se reemplaza con hueso displásico penetrado por tejido fibroso. La lesión del hueso que produce es una mezcla casual de tejido fibroso inmaduro y fragmentos pequeños de hueso trabecular inmaduro.
Se le puede clasificar en tres grupos: el tipo monostótico en que hay ataque de un solo hueso; la forma poliostótica, en que hay ataque de múltiples huesos, y una forma poliostótica acompañada de anormalidades endocrinas, como pubertad precoz, maduración esquelética prematura o hipertiroidismo, conjunto conocido actualmente como enfermedad de Albright.
Causas
No se ha precisado la causa exacta ni la naturaleza
fundamental de la displasia fibrosa. Al parecer es una anormalidad del
mesénquima osteógeno, durante el desarrollo. El tejido fibroso progresivo
prolifera dentro de la médula ósea, comprime la corteza desde el interior, y
produce expansión.
Se ha sugerido una posible base congénita, por la afectación unilateral en la
forma poliostótica de la enfermedad. La imagen histopatológica señala que las
formas monostótica y poliostótica son manifestaciones del mismo proceso
biológico. Las pruebas existentes anulan la posibilidad de una disfunción
endocrina como causa de las lesiones esqueléticas.
El trastorno no es hereditario, y todos los casos publicados son esporádicos.
Firat y ¡ Stutzman publicaron dos casos en una madre y su hija. Lemli señaló el
caso de gemelas monocigóticas, una con el síndrome de McCune-Albright y la otra
sólo con signos esporádicos de displasia fibrosa en el cráneo. Al parecer, la
expresión fenotípica varía a pesar de genotipos idénticos y la aparición de
displasia fibrosa en gemelos monocigóticos sugiere la posibilidad de un origen
genético.
Edad y sexo
Ocurre
típicamente
en
la
adolescencia,
aunque
una
cuarta
parte
de
las
lesiones
se
dan
en
los
adultos.
Sitios
afectados:
Los sitios más comunes de afectación monostótica incluyen las partes proximales del fémur, tibia, húmero, costillas, cráneo, y la columna cervical, mandíbula y las costillas (muy frecuente).
Fractura
patológica:
El
hueso
se
deforma
por
múltiples
fracturas
de
estrés
que
puede
llevar
en
el
futuro
a
una
fractura
patológica.
Las
fracturas
de
estrés,
muy
dolorosas,
son
especialmente
comunes
en
el
cuello
femoral.
Aunque
el
hueso
displásico
cura
después
de
la
fractura,
el
callo
resultante
también
es
displásico,
y
la
enfermedad
persiste.
Condiciones
asociadas:
Síndrome
de
Albrights: Pubertad
precoz
y
pigmentaciones
café
con
leche
en
las
mujeres.
El
niño
con
displasia
fibrosa
monostótica normalmente
no
tiene
ningún
síntoma.
Las
lesiones
poliostóticas
que
normalmente
se
presentan
mas
precoces
pueden
ser
unilaterales
o
generalizadas,
afectando
a
los
huesos
largos,
manos,
pies,
y
pelvis;
referencias:
Some
aspects
of
polyostotic
fibrous
dysplasia:
Possible
hypothesis
to
account
for
the
associated
endocrine
changes.
Dedifferentiated
chondrosarcoma
in
Albright
syndrome. A
case
report
and
review
of
the
literature.
|
|
|
|
|
|
|
|
|

Presentación
clínica:
Las
lesiones
pueden
ocurrir
en
un
hueso
(monostótica)
o
en
muchos
(poliostótica).
De
vez
en
cuando,
un
niño
puede
presentarse
con
fractura
patológica
o
deformidad
angular.
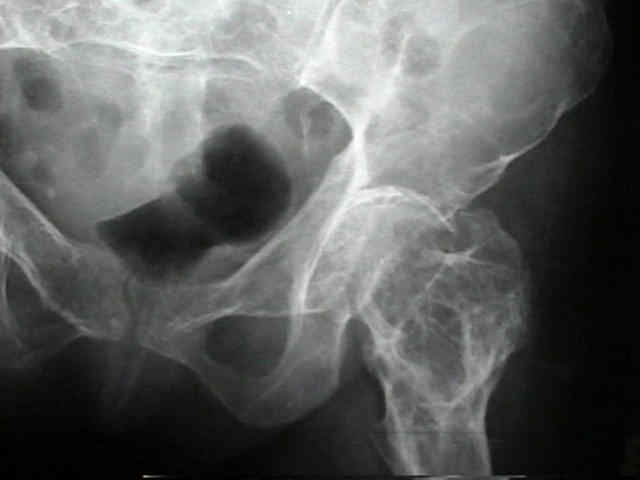
Displasia fibrosa monostótica
Normalmente afecta al fémur, especialmente al cuello, la tibia, las costillas y la base del cráneo. Nace en la parte central del hueso. Normalmente está separada del cartílago de crecimiento en los niños y de la superficie articular en los adultos.
Esta lesión es asintomática y generalmente es descubierta por otras razones.
En algún caso hay dolor, tumefacción y deformidad.
La complicación más frecuente es una fractura, y a menudo es el síntoma inicial principalmente en extremidades inferiores.
Las lesiones craneales pueden progresar lentamente hasta la vida adulta y afectar al nervio óptico o las estructuras del oído medio.
Displasia
fibrosa
poliostótica:
Se
presentan
en
niños
de
alrededor
de
10
años
de
edad.
La distribución y extensión de las lesiones varia ampliamente, desde el compromiso de unos pocos huesos de una extremidad hasta la afectación de más del 50% de los huesos del esqueleto, en el 90% de los casos unilateral.
Frecuentemente está implicada la pelvis, seguida delos huesos largos, cráneo, costillas, y extremidad proximal del fémur.
Aunque las lesiones son similares a la forma monostótica, tienen una apariencia más agresiva, y la enfermedad progresa rápidamente, hasta que el esqueleto alcanza su madurez; después solo un 5% de las lesiones sigue creciendo.
A
diferencia
de
la
forma
monostótica,
la
displasia
fibrosa
poliostótica
da
síntomas.
Los
hallazgos
incluyen
dolor,
fractura
patológica,
cojera,
o
deformidad
del
miembro.
La asociación de tumores fibrosos y fibromixomatosos de los tejidos blandos con displasia fibrosa poliostótica se conoce como síndrome de Mazabraud.
La complicación más frecuente de la displasia fibrosa poliostótica es la fractura patológica
En
general
las
anormalidades
óseas
se
presentan
antes
de
la
edad
20
años;
Estudios
de
imagen:
Radiografías
La
apariencia
de
las
lesiones
es
inconstante
y
depende
de
la
proporción
de
los
componentes
óseos
y
fibrosos
de
la
lesión
que
ocupan
grandes
áreas
en
el
interior
del
hueso.
Si
predomina
el
componente
de
tejido
conectivo
se
verá
una
lesión
diafisaria
intramedular
radiolúcida
que
se
combina
con
adelgazamiento
y
abombamiento
de
la
cortical
cortical.
En el nivel de lesión suele haber deformidad angular del hueso.
La
lesión
activa
puede
progresar
en
el
tamaño
y
deformidad.
lesión
tipo
quístico:
Radiolucente
con
un
margen
reactivo,
ninguna
trabécula,
y
el
espesor
cortical
normal.
lesiones
tipo
pagetoide:
Patrón
trabecular
más
denso
que
el
hueso
normal.
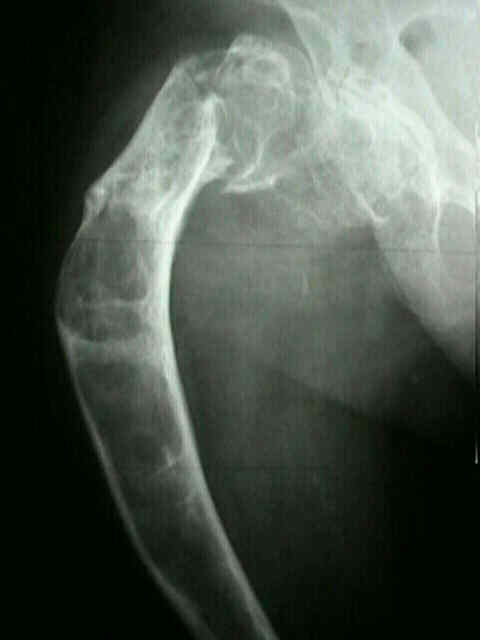
deformidad
en
cayado
o
bastón:
La
amplia
afectación
del
fémur
proximal
produce
una
característica
deformidad
en
varus
que
se
parece
a
la
curva
de
un
bastón.
Gammagrafía:
Es la forma más rápida para determinar la distribución de las lesiones esqueléticas, además de permitir descubrir lesiones en sitios insospechados.
Revela
intensa
captación
del
radioisótopo
que
refleja
la
magnitud
del
tumor
vista
en
la
radiografía,
especialmente
cuando
la
lesión
es
activa.
TAC y RNM:
Pueda
ayudar
evaluar
con
exactitud
la
magnitud
de
la
afectación
ósea.
La intensidad de señal en la RNM es moderadamente baja en T1, mientras en T2 es alta o media. Con gadolinio, la mayoría de las lesiones muestran un incremento central de contraste y algunos anillos periféricos. En general, la intensidad de señal depende de la cantidad de trabéculas óseas, colágeno, quistes y hemorragias.
Histología:
Hay
una
colección
irregular
de
trozos
pequeños
de
tejido
inmaduro
trabecular
rodeado
por abundante
proliferación
fibroblástica
de
matriz
de
tejido
fibroso
y
tejido
trabecular
inmaduro.
Algunas
lesiones
pueden
contener
cantidades
grandes
de
cartílago
benigno,
mientras
otros
pueden
tener
componentes
quísticos
grandes.
Histológicamente,
los
rasgos
típicos
incluyen
hueso
trabecular
displásico
truncado
produciendo
segmentos
cortos
del
hueso,
irregulares
(llamados
sopa
del
alfabeto
chino).
Las
trabéculas
displásicas
están
típicamente
presentes
dentro
del
estroma
fibroso
que
reemplaza
el
hueso
normal,
y
la
actividad
celular
de
estas
lesiones
es
moderada.
La
apariencia
se
ha
asemejado
a
sopa
de
letras,
con
trabéculas
que
se
parecen
a
"Ces,
Oes, Us".
Trabéculas:
-
aparecen
inmaduras;
-
no
están
revestidas
con
osteoblastos
(como
en
el
fibroma
osificante);
-
no
contiene
las
líneas
de
cemento;
-
no
remodela
según
la
tensión;
-
el
estroma
fibroso
es
desorganizado
y
reemplaza
a
la
médula
normal;
Diagnóstico
diferencial:
Radiográfico
Si la lesión de la displasia fibrosa contiene cartílago y muestra calcificaciones visibles puede entrar en el diagnóstico diferencial con el encondroma si la lesión es solitaria o con la encondromatosis si la lesión es poliostótica.
En algunas ocasiones , si la lesión es solitaria puede recordar al fibroma desmoplástico.
Si se presenta como un foco solitario en la tibia, las posibilidades diagnósticas son displasia osteofibrosa y adamantinoma.
En las fases precoces de evolución, particularmente en las lesiones radiolucentes situadas en el húmero proximal puede confundirse con un quiste óseo simple.
Las lesiones que expanden la cortical pueden recordar a un quiste óseo aneurismático. En estos casos la lesión es excéntrica.
Una lesión monostótica con anillo escleroso en el húmero proximal se puede confundir con un infarto óseo intramedular.
La displasia fibrosa poliostótica entrará en el diagnostico diferencial con la encondromatosis y la neurofibromatosis. La encondromatosis a diferencia de la displasia fibrosa poliostótica, las lesiones pueden extenderse en la parte articular del hueso y se extienden bandas radiolucentes del platillo de crecimiento a la metáfisis. En la neurofibromatosis la afectación esquelética, normalmente, se ven deformidades en los huesos largos sin los cambios típicos intra-medulares de la displasia fibrosa poliostótica. Si la neurofibromatosis se asocia con fibromas no osificantes múltiples (síndrome de Jaffe-Campanacci) puede confundirse con la displasia fibrosa.
Patológico
Osteosarcoma fibroblástico y fibrosarcoma. Las células del osteosarcoma y su núcleo son diferentes y las mitosis son frecuentes. La formación ósea del tumor es más amorfa, sin estructura trabecular distintiva. No obstante el osteosarcoma central de bajo grado puede suponer un problema para el patólogo.
El adamantinoma puede recordar a la displasia cuando se acompaña de abundante tejido fibroblástico, no obstante las células epiteloides son características y diagnósticas.
Cuando la formación de hueso es limitada, puede ser problemático el diagnóstico diferencial con el fibroma no osificante. En estos caso puede ayudar la ausencia de grupos de pseudo osteoclastos (células gigantes) y hemosiderina.
En algunos casos también pueden entrar en el diagnóstico diferencial el hiperparatiroidismo, el osteoblastoma y la enfermedad de Paget.
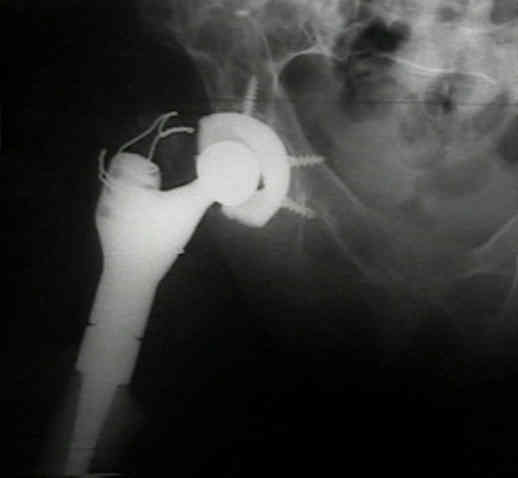
Tratamiento:
El
tratamiento
de
la
displasia
fibrosa
sigue
siendo
una
tarea
desafiante
en
niños
y adultos.
El
paciente
típico
que
tiene
una
lesión
monostótica
grande,
dolorosa
o poliostótica
normalmente
se
beneficia
de
la
fijación
intramedular
de
ese
segmento
óseo.
Se
requiere
una
biopsia
en
la
mayoría
de
las
situaciones.
El
curetaje
se
asocia
con
una
proporción
muy
alta
de
repetición
local
y
por
consiguiente,
normalmente
no
se
recomienda.
Así,
se
tratan
típicamente
mejor
las
lesiones
de
displasia
fibrosa
con
biopsia
seguida
por
algún
tipo
de
injerto
cortical
o
fijación
del
implante
para
estabilizar
el
segmento
del
hueso
largo.
En
el
fémur,
esto
se
logra
con
un
injerto
cortical
de
peroné o
un
clavo
femoral
encerrojado,
dependiendo
de
la
edad
del
paciente.
El
niño
con
una
lesión
grande
tendrá
una
deformidad
dolorosa
progresiva,
continuada,
y
pueden
requerirse
intervenciones
múltiples.
Esto
es
sobre
todo
cierto
para
los
pacientes
con
enfermedad
severa.
Consideraciones
preoperatorias:
Observe que los adultos pueden experimentar hipoxemia intra-operatoria y desvíos durante la cirugía (debido a shunts AV), y que estos pacientes pueden beneficiarse intraoperatoriamente de la monitorización.
La
meta
del
tratamiento
es
la
prevención
de
la
deformidad
y
la
fractura.
El
tratamiento
de
la
displasia
fibrosa
implica
el
fortaleciendo
y
enderezamiento
del
hueso
afecto.
La
resección
y
el
curetaje
no
están
indicados,
ya
que
el
hueso
displásico
normalmente
recidiva.
La
proporción
de
recurrencia
con
el
curetaje
simple
e
injerto
de
hueso,
especialmente
en
el
grupo
de
edad
pediátrico
es
alto.
Las
lesiones
grandes
con
deformidad
angular
son
enderezadas
mejor
e
injertadas
con
hueso
cortical.
-
El
hueso
cortical
probablemente
es
menos
resorbido
que
el
esponjoso
y
más
probablemente, proporcionara
un
apoyo
estructural
permanente.
-
Este
es mejor
realizarlo
usando
autoinjertos
de
hueso
cortical
(tomado
del
peroné),
qué
mínimamente
remodela
después
de
la
incorporación.
-
El
injerto
con
hueso
cortical
es
particularmente
apropiado
para
el
manejo
de
las
lesiones en
las
áreas
de
tensión
altas
como
el
cuello
femoral.
-
Los
métodos
de
tratamiento
alternativos
son
la
reconstrucción
con
aloinjerto
de
hueso
cortical
o
fijación
con
un
clavo
intramedular;
Extremidad
inferior:
La deformidad en coxa vara progresiva es común qué puede llevar en el futuro a la deformidad en cayado.
La
inmensa
mayoría
de
pacientes
con
lesiones
poliostóticas
tendrá
afectación
del
fémur
proximal
y
en
la
mayoría
de
éstos
pacientes,
habrá
afectación
más
frecuente
y
más
severo
del
calcar
de
manera
que
será
incapaz
de
soportar
una
fijación
interna.
Las
indicaciones
para
la
cirugía
incluyen
la
deformidad
progresiva,
dolor
persistente,
y
fracaso
de
tratamiento
conservador.
La
lesiones
con
riesgo
de
fractura
patológica
son
premonitorias
en
lesiones
líticas
de
2.5
centímetros
(o
mayor)
que
afecta
el
fémur
del
proximal.
La
osteotomía
valguizante
y
fijación
interna:
se
realiza
mejor
en
la
enfermedad
temprana.
La osteotomía de desplazamiento medial se usa cuando hay implicación del calcar o cuando hay un hueso de baja calidad.
La
ventana
cortical
se
usa
para
la
extirpación
del
tejido
fibroso
del
tumor
y
para
el
injerto
del
hueso.
Extremidad
superior:
Las fracturas patológicas de la extremidad superior pueden ser tratadas por métodos cerrados en contraste con las de la extremidad inferior que se fijarán internamente.
Tratamiento no quirúrgico
El tratamiento médico puede incluir el pamidronato disódico, que está indicado, sobre todo, en el tratamiento de la hipercalcemia en pacientes con enfermedad de Paget y neoplasias malignas. El Pamidronato no está indicado en el tratamiento de la displasia fibrosa, pero los estudios recientes (ensayos clínicos preliminares) han demostrado que las infusiones múltiples de esta droga pueden aliviar el dolor óseo asociado a la enfermedad y en algún caso mejoran el aspecto radiográfico. El ejercicio es importante para evitar el aumento del peso y preservar la movilidad articular.
El tratamiento con radioterapia no se ha mostrado eficaz en la erradicación de la enfermedad y puede conducir a la transformación maligna.
Pronóstico
En resumen:
La
progresión
de
la
enfermedad
es
a
menudo
impredecible.
Las
lesiones monostóticas:
tienen
buen
pronóstico.
Las
lesiones poliostóticas: tienden
a
permanecer
más
activas
o
agresivas.
Ocasionalmente
sufren
una
transformación
maligna
a
osteosarcoma
o
fibrosarcoma.