... Sarcomas de Partes Blandas ...
- Introducción
- Incidencia
- Etiología
- Clasificación
- Presentación clínica
- Distribución anatómica
- Evaluación diagnóstica
Introducción
Los sarcomas de tejidos blandos son tumores heterogéneos. Aunque su frecuencia es muy rara, es de las neoplasias mas raras, solo implica el 1% de todas las neoplasias. La derivación del tejido neoplásico esta relacionado con el mesénquima de los órganos (tejido conectivo: ligamentos, tendones, músculos, tejido adiposo y vaina de Schwann). En base a esto, este tipo de neoplasia puede encontrarse en cualquier parte del tejido humano ya que todos los órganos tienen mesénquima. Es sumamente raro pero implica una de las condiciones con mayor agresividad en neoplasias malignas. Se les llamó sarcomas y etimológicamente quiere decir carne de pescado, son células mesodérmicas que dan origen a estroma orgánico.
Como un indicador de la dificultad en el desarrollo de un entendimiento profundo de la historia natural y los patrones de respuesta a las diversas series de estrategias de tratamiento empleadas, hay que considerar el número tan pequeño de sarcomas que pueden encontrarse en un solo sitio anatómico. Hay sólo aproximadamente 500 liposarcomas del muslo diagnosticados por año en los Estados Unidos comparados al número esperado de aproximadamente 182,000 pacientes diagnosticadas con carcinoma de mama en el año 2000. Así, el número de adenocarcinomas de un sitio anatómico en las mujeres excede, por un factor de aproximadamente 22, el número de sarcomas de tejidos blandos de todas las variedades patológicas, en todos los sitios anatómicos, todas las edades y ambos sexos.
El diagnóstico de una sarcoma tiene un impacto negativo en el estado de salud y calidad de vida de un paciente. Aproximadamente 50 por ciento de pacientes sucumben finalmente a su enfermedad y el tratamiento es a menudo asociado con morbosidad aguda y larga evolución. Debido a la rareza y gravedad de estos tumores, seria recomendable que se evaluaran y trataran en un centro con un equipo de subespecialistas cuya práctica se limita a los sarcomas. Estos subespecialistas incluirían a médicos cirujanos, cirujanos ortopédicos, oncólogos, pediatras, y radioterapeutas.
Los beneficios que se proporcionan al paciente al ser evaluados por un “equipo de sarcoma” previo al tratamiento definitivo incluyen la planificación de la biopsia por un especialista. Una biopsia incorrectamente hecha puede comprometer la actuación de la cirugía definitiva o administración de la dosis de radiación adecuada. Los procedimientos de diagnóstico especiales en el tejido fresco pueden hacerse con garantía. Se minimizan demandas para los estudios radiológicos, se evita la duplicación, y se reduce el tiempo de aplicación del protocolo terapéutico definitivo.
La especialización ganada de la concentración de tiempo y esfuerzo en este tumor raro por los subespecialistas debe proporcionar un poco de ganancia en el resultado final. Esto se ilustra por un estudio que examinó el resultado en 375 pacientes con sarcoma de tejidos blandos de las extremidades y torso según el tiempo de referencia a un centro de tumores en la Región Sur de Suecia. Comparó a los pacientes referidos preoperatoriamente, se vio que aumentaron las recurrencias locales en pacientes no referidos nunca a (2.4 veces superior) y en pacientes referidos después de la cirugía (1.3 veces superior).
Incidencia
Son tumores poco comunes y por su origen diverso son numerosos considerando
su histopatología. La estimación actual es que representa aproximadamente un
0.87 por ciento de los nuevos tumores malignos. De
estos
tumores,
76
por
ciento
surgen
en
el
tejido
blando
y
el
resto
en
el
hueso.
La incidencia ha aumentado durante las últimas décadas. Pueden presentarse en cualquier lugar del cuerpo, sin embargo
el 50% se localiza en las extremidades, 15% en miembros superiores, 37% en
miembros inferiores, intra-abdominal en 14%, viscerales 13%, tronco 9%, torácicos
5%, cabeza y cuello 5%.
Los sarcomas de partes blandas constituyen el 1% de cáncer humano y es más
frecuente sobre los 50 años, pero no hay una edad especifica ya que puede
aparecer a lo largo de toda la vida. La relación hombre mujer es de 1.2 a 1 pero
algunos autores indican que no hay una diferencia significativa. El 90% de todas
estas neoplasias son en pacientes blancos, 6% en negros, 4% en otras razas.
Etiología
Existe la evidencia para sugerir que puede haber una predisposición genética en el desarrollo de sarcomas. La neurofibromatosis se sabe que es un precursor de sarcomas de los tejidos blandos y el sarcoma se asocia con el síndrome de Li-Fraumeni o síndrome de SBLA; este síndrome es una condición poco frecuente, autosómica dominante caracterizada por neoplasias mesenquimales y epiteliales en múltiples lugares del organismo. La mutación del gen supresor p53, aparentemente predispone a los miembros de la familia afectados hereditariamente a desarrollar ciertos canceres (sarcoma, mama, hueso, cerebro, pulmón, laringe, leucemia, corteza suprarrenal); estos pacientes tienen 30 veces más riesgo de desarrollar un sarcoma cuando se compara con la población general. Y la investigación ha mostrado una relación entre las anormalidades del regulador p53 y RB1 del ciclo celular y los sarcomas.
También se sabe que la exposición a la radiación durante el tratamiento para otras enfermedades predispone al desarrollo de sarcomas. Esto es particularmente evidente en pacientes tratados de cáncer de mama o linfoma. El linfedema crónico (síndrome de Stewart-Treves) se asocia con desarrollo de linfangiosarcoma. Pacientes que sufren linfangiosarcoma son típicamente sobrevivientes de cáncer de mama que tenían mastectomías radicales seguidos por radioterapia, resultando el linfedema crónico en la extremidad superior. El linfangiosarcoma no sólo se desarrolla en el campo de la radiación, sino que también lo hace en áreas no irradiadas.
Es interesante destacar que el síndrome de Stewart-Treves ocurre, también, tras una obstrucción parasitaria, indicando un papel para el linfedema crónico así como la radiación en el desarrollo de linfosarcoma. El riesgo de desarrollar angiosarcoma después de sufrir un cáncer de mama tratado con radioterapia aumenta en 60 veces.
Los sarcomas se asocian estadísticamente con la exposición química, como las dioxinas y PVCs, trauma, y hemocromatosis. La vía causal real, sin embargo, permanece incierta.
El traumatismo no está relacionado con los sarcomas, el único tumor de partes
blandas posiblemente ligado al trauma es el desmoide.
Clasificación
Muchos sarcomas se clasifican histopatológicamente según sus características de diferenciación y, por consiguiente, por su tejido presunto de origen. Los ejemplos más comunes incluyen fibrohistiocitoma maligno, liposarcoma, sarcoma sinovial, leiomiosarcoma, rabdomiosarcoma, fibrosarcoma y angiosarcoma. Para otros sarcomas, la designación refleja el patrón histológico, ejemplo, el sarcoma alveolar de partes blandas, sarcoma epiteloide, y el sarcoma de células claras. Se piensan que los sarcomas aparecen de novo en casi todos los casos; normalmente no se desarrollan de neoplasias benignas preexistentes.
Hay también tumores de tejidos blandos benignos. La incidencia relativa de tumores benignos a los tumores malignos es aproximadamente 100 :1. Los ejemplos incluyen desmoides, lipoma atípico, y neuroma. Hay lesiones no neoplásicas que pueden confundirse con lesiones benignas o con neoplasias mesenquimales de bajo grado, (ejemplo, la contractura de Dupuytren, la fibromatosis plantar).
Tumores y pseudotumores de partes blandas |
||
| Tipo del tejido | Benigno | Maligno |
| Óseo | Miositis osificante |
Osteosarcoma Extraóseo |
Cartilaginoso |
Condroma Condromatosis sinovial |
Condrosarcoma mixoide extraóseo Cond. mesenquimatoso extraóseo |
| Fibroso | Fibroma Fibroma desmoplástico Fibromatosis -Queloide -Fibromatosis por irradiación -Fascitis palmar y plantar -Enfermedad de Peyronie -Tortícolis congénito (fib. colli) -Fibroma aponeurótico juvenil -Elastofibroma dorsal -Fibromatosis progresiva. -Fib. generalizada congénita -Fib. dérmica infantil -Hamartoma fibro. de la lactancia -Fib. difusa infantil -Fib. infantil agresiva |
Fibrosarcoma Dermatofibrosarcoma protuberans (porminenete) |
| Fibrohistiocítico | Histiocitoma fibroso benigno o Dermatofibroma |
Histiocitoma fibroso maligno |
| Sinovial | Sinovitis Villonodular Pigmentada Ganglión |
|
| Vascular | Hemangioma Hemangiopericitoma Hemangioendotelioma |
Angiosarcoma Linfangiosarcoma Hemangiopericitoma Maligno Hemangioendotelioma malig. |
| Graso | Lipoma |
Liposarcoma |
| Nervioso | Neurilemoma Neurofibroma Neuroblastoma |
Neurosarcoma Neurofibrosarcoma
|
| Muscular | Leiomioma Rabdomioma |
Leiomiosarcoma Rabdomiosarcoma |
| Piel/Dermis | Melanoma maligno (MMSP)* Carcinoma celular escamoso |
|
| Más de un tipo de tejido | Mesenquimoma fibrocartilaginoso |
Mesenquimoma maligno Tumor triton maligno |
| Desconocido | TCG de Vaina del Tendón Mixoblastoma de células gigantes Mixoma de las partes blandas Esbozo retiniano |
Sarcoma epiteloide Sarcoma alveolar de partes blandas Sarcoma de Ewing Tumor desmoplasico de células redondas pequeñas. |
| *Melanoma maligno de partes bandas (sarcoma de células claras) | ||
Presentación
clínica
El paciente con un sarcoma del tejido blando se presenta frecuentemente con una tumoración sin dolor de unas semanas o unos meses de duración. Menos común es la presentación con dolor o síntomas secundarios a los efectos de la compresión de un nervio o hueso por una masa ignorada. Las metástasis en la presentación inicial son raras. Esto se ilustró en un estudio de todos los pacientes diagnosticados de sarcoma en el Distrito Del sur de Suecia; sólo 19 de 278 (7 por ciento) tenían enfermedad metastática en el momento de diagnóstico inicial.
Distribución
anatómica
Los sarcomas del tejido blando ocurren en todos los sitios anatómicos del cuerpo. La frecuencia no parece ser una función simple de la abundancia de este tipo de tejido. Como un ejemplo, el liposarcoma no es común en los depósitos grasos grandes del abdomen; el muslo es un sitio frecuente dónde el liposarcoma se levanta a menudo profundamente en la masa del músculo, en lugar de en la grasa hipodérmica.
Cierto tipos histológicos de sarcoma de tejidos blandos difieren en su distribución anatómica. Sólo 14 por ciento de todos los sarcomas de tejidos blandos se presentan en la extremidad superior; en comparación, 40 a 50 por ciento de todos las sarcomas epiteloides aparecen en el antebrazo y dedos.
La distribución anatómica de sarcomas de tejidos blandos en 4508 pacientes repasados por el American College of Surgery fue:
- Muslo, nalga, y región de la ingle - 46 por ciento
- Extremidad Superior - 13 por ciento
- Cabeza y región del cuello - 9 por ciento
- Torso - 18 por ciento
- Tejidos del retroperitoneo - 13 por ciento
Los sarcomas del tejido blando crecen por extensión local directa, infiltrando tejidos y estructuras adyacentes, de vez en cuando con skip áreas. Generalmente se extienden a lo largo de los planos del tejido y raramente atraviesan o invaden el plano fascial mayor o el hueso.
La diseminación a los ganglios regionales es poco frecuente salvo el rabdomiosarcoma, sarcoma epiteloide, y los sarcomas vasculares. En una serie de 323 pacientes, las metástasis a los ganglios linfáticos regionales o al diagnóstico o como el primer sitio de metástasis, se vio en aproximadamente 6 por ciento de pacientes. La frecuencia de implicación de adenopatía regional estaba en función del grado: grado 1, 0 por ciento,; grado 2, 2.5 por ciento,; y grado 3, 12 por ciento. Entre los pacientes con sarcomas de grado 3, el riesgo era más grande en aquellos con rabdomiosarcoma (cinco de 14) y sarcoma epiteloide (cuatro de cinco). La afectación ganglionar era rara en los pacientes con las lesiones <5 centímetro de diámetro.
Los sarcomas del tejido blando son caracterizados a menudo por el crecimiento local e invasión. Aunque las metástasis en el momento de la presentación son infrecuentes, después hay diseminación a distancia frecuente en el curso de la enfermedad. La incidencia de metástasis a distancia es una función del grado y tamaño del sarcoma como ilustran las observaciones siguientes:
|
|
En un informe de 278 pacientes, el tiempo del medio al desarrollo de metástasis a distancia en los pacientes con sarcomas grados 1 a 2, 3, y 4 eran respectivamente 27, 22, y 12 meses. |
|
|
En un estudio de 501 pacientes que lograron control local de su sarcoma de tejidos blandos de grado 2 a 3, la incidencia a 5 años de metástasis a distancia aumentó de 3 por ciento a 55 a 60 por ciento en función de si el diámetro del tumor era de 2.5 centímetro o menos a >20 centímetro (vea tabla I). Este efecto del tamaño en la frecuencia de metástasis a distancia se ha observado para otros tumores, como el carcinoma de mama. |
Tabla I: Probabilidad de metástasis a distancia a los 5 años en 501 pacientes tratados localmente con radioterapia y cirugía en función del tamaño del tumor para los grados 2 y 3. |
||
| Tamaño en mm | Número de pacientes | Metástasis a distancia, % |
> 26 25 - 50 51 - 100 101 - 150 151 - 200 < 200 Total |
58
128 177 68 49 21 501 |
3
22 34 43 58 57 35 |
| Koscielny, S, Tubiana, M, Le, MG, et al. Breast cancer: relationship between the size of the primary tumour and the probability of metastatic dissemination. Br J Cancer 1984; 49:709 | ||
El sitio primario de metástasis de los sarcomas de las extremidades es el pulmón en 70 a 80 por ciento de pacientes que desarrollan metástasis a distancia. La excepción principal es el liposarcoma que frecuentemente metastatiza a otros sitios del tejido blando. Sin embargo, el pulmón es finalmente implicado en, virtualmente, todos los paciente que desarrollan metástasis, y ésta es la causa predominante de muerte.
Evaluación
diagnóstica
Una historia completa y el examen físico deben definir las áreas anatómicas implicadas, y el paciente debe evaluarse para la evidencia de invasión de la piel, vasos mayores, nervios o del hueso, el estado de ganglios linfáticos regionales, y la presencia de edema. Los estudios radiográficos proporcionan los medios más buenos de evaluar el sitio anatómico, la magnitud de invasión local, y el modelo de extensión. La biopsia proporciona la base del diagnóstico definitivo.
La meta primaria es determinar si la masa es benigna o maligna.
Previamente a la biopsia el médico debe determinar:
|
|
si la masa está arriba o debajo de la fascia (las lesiones profundas son más a menudo malignas); |
|
|
las excepciones son fibrohistiocitoma maligno y leiomiosarcoma que se pueden dar en los tejidos subcutáneos; |
|
|
tamaño de la masa: |
1. si la masa es mayor de cinco centímetros, especialmente si es profundo y en el muslo, es probable que sea un sarcoma.
2. si hay cambios visibles en la radiografía (como calcificación del tejido blando que puede verse en el sarcoma sinovial).
Se ha recomendado por D. Springfield M.D. (JBJS 1996) que las masas superficiales que se sienten como grasa (lipoma), qué no causan dolor y qué no ha cambiado de tamaño puede seguirse clínicamente; las masas que son dolorosas, han cambiado de tamaño, o que los resultados de la radiografía sospechosos, requerirá RM y biopsia (biopsia incisional o por aguja).
Estudios
Radiográficos
El estudio de la radiografía simple del sitio afectado es apropiado para evaluar el estado del hueso adyacente. Además, la tomografía computadorizada (TAC) y la resonancia magnética (RM) son decisivas en la determinación de la localización del tumor con relación a los planos fasciales, huesos, vasos, nervios y órganos. Las dimensiones globales del tumor pueden estimarse en estas imágenes para el cómputo de volumen del tumor; sin embargo, estas imágenes pueden dar una sobrestimación del tamaño de la lesión.
RM y TAC. La RM generalmente es la técnica más valiosa de imagen; sin embargo, la TAC es importante en la predicción de destrucción del hueso cortical, y es superior para la valoración de lesiones en el parénquima pulmonar. La demostración de necrosis del tumor en TAC es un indicador de prognosis pobre. En un estudio con tres años de seguimiento, las metástasis se desarrollaron en 19 de 41 pacientes con necrosis comparada a sólo uno de 10 pacientes sin este hallazgo, incluso controlando el tamaño del tumor. La necrosis es rara en los tumores de grados bajos.
En
el
paciente
recientemente
diagnosticado
con
un
sarcoma
del
tejido
blando,
se
debe
hacer
como
rutina
una TAC
de
tórax
para
examinar
las
metástasis
a
distancia.
Este
estudio
sirve
como
una
valoración
básica
de
los
pulmones.
En
los
exámenes
de
seguimiento,
pueden
alternarse
las
radiografías
simples
de
tórax
con
el
examen TAC.
Para
los
pacientes
con
sarcoma
del
retroperitoneo,
el
hígado
debe
ser
preoperatoriamente
inspeccionado
ya
que
puede
ser
el
primer
sitio
de
metástasis.
También
debe realizarse TAC
abdominal
en
los
pacientes
con
el
liposarcomas
de
la
extremidad
ya
que
estos
tumores
pueden
metastatizar
al
retroperitoneo
y
al
hígado.
PET Scan. La PET Scan con fluorodeoxyglucosa (el FDG-PET) es una técnica cuantitativa con una esperanza sustancial en el estadiaje y evaluación de sarcomas de los tejidos blandos. La PET Scan, que mide la actividad metabólica de la glucosa, puede descubrir extensión local insospechada o diseminación a distancia. En un informe, la sensibilidad de la PET Scan para el descubrimiento de sarcoma del tejido blando era del 100 por ciento.
La PET también puede distinguir las lesiones benignas y la lesiones de alto grado; sin embargo, su capacidad para diferenciar lesiones benignas y de bajo grado a intermedio está limitada. La sensibilidad para los sarcomas de grado bajo parece ser menor que para el grado intermedio o los tumores de grado alto. Además, la magnitud de la reducción en la actividad metabólica de la glucosa puede usarse para estimar el grado de necrosis inducido por la radiación y quimioterapia, proporcionando un método no invasivo de evaluar la respuesta.
Rastreo óseo. El RO normalmente no es útil para el estadiaje inicial. En ausencia de metástasis múltiples en otros sitios, las metástasis del hueso son raras en los adultos, salvo el liposarcoma. Además, un RO positivo en presencia de un tumor del tejido blando adyacente al hueso no es evidencia buena de invasión del hueso; este diagnóstico se hace mejor por la demostración de pérdida de hueso cortical en radiografías simples o TAC con ventanas del hueso.
Biopsia
La biopsia es un paso decisivo en el manejo del paciente con sarcoma de tejidos blandos. Sin embargo, la actuación óptima de la biopsia puede ser difícil y debe hacerse por cirujanos experimentados. La biopsia diagnóstica inicial de una masa de tejidos blandos debe tener en cuenta los requisitos para el futuro definitivo quirúrgico y o procedimientos de la radiación. Cada uno de éstos puede comprometerse gravemente por una técnica o localización de la biopsia inadecuada (vea debajo).
La biopsia puede realizarse por la técnica incisional, Tru-cut, la aspiración con aguja fina (PAAF), o, en circunstancias especiales, escisión. El procedimiento más frecuente es la biopsia TRU-CUT que generalmente proporciona tejido adecuado para el establecimiento del tipo histológico y el grado. Las ventajas son la facilidad, disponibilidad (qué minimiza los retrasos), la morbosidad mínima, y el costo bajo. Sin embargo, cuando se plantea la citometría de flujo, citogenética, el análisis genético, y/o el cultivo in vitro, se prefiere una biopsia incisional normalmente para proporcionar el material suficiente. La muestra más grande obtenida por la biopsia incisional le proporciona una base más segura al patólogo para llegar al diagnóstico, en parte debido al grado de heterogeneidad de la morfología a lo largo del tumor.
Biopsia Incisional. La biopsia Incisional de una sarcoma de tejidos blandos debe situarse apropiadamente, de corta longitud (sólo unos centímetros, exceptuando lo raros sarcomas situados profundamente), y, para lesiones localizadas en las extremidades, en el eje largo. La incisión no debe ponerse encima de las prominencias óseas, como la espinilla de la tibia o el cóndilo femoral. Se puede realizar una biopsia incorrectamente si hay una inadecuada valoración radiológica prequirúrgica.
Una biopsia inicial hecha deficientemente puede imposibilitar o puede complicar la resección quirúrgica subsiguiente, la preparación de flaps, y/o la reparación cosmética. En tales pacientes, se requiere una escisión quirúrgica más ancha en el momento de la cirugía definitiva para lograr los márgenes limpios que habría sido necesario si una biopsia incisional apropiada se hubiera realizado en el procedimiento inicial. En dos series, 18 a 19 por ciento de pacientes tenían que sufrir una intervención más compleja o recibir quimioterapia adyuvante o radioterapia debido a la actuación de una biopsia incorrecta.
Durante la biopsia incisional, debe prestarse atención meticulosa a la hemostasis. Se debe presumir que las áreas de equimosis alrededor del sitio de la biopsia pueden contener células del tumor. Las áreas extensas de equimosis hacen que la irradiación o rereseccion sea más difícil.
La biopsia con TRU-CUT - la biopsia con Tru-Cut es muy exacta en el diagnóstico de tumores de tejidos blandos malignos. Esta biopsia (biopsia con Tru-Cut) evita las complicaciones de la biopsia abierta y proporciona ventajas importantes en cuanto a la reducción del costo y del tiempo. En un estudio de 60 biopsias con Tru-Cut, el 93 por ciento de las muestras eran adecuadas. De las muestras adecuadas, el diagnóstico histológico final a partir de la resección definitiva puesta en correlación con el diagnóstico de la biopsia con Tru-Cut para la presencia de malignidad, grado, y subtipo histológico en 95, 88, y 75 por ciento, respectivamente. Las muestras de biopsia de sección congeladas, fueron adecuadas para el diagnóstico de malignidad, pero no era suficientemente exacto para el grado y el subtipo histológico.
Punción Aspiración con Aguja Fina (PAAF) es muy exacto para el diagnóstico de sarcomas del tejido blando, pero normalmente no permite la distinción de subtipo histológico o el grado. En una serie de 95 pacientes, no había ningún resultado falso positivo, 5 por ciento resultados falsamente benignos, y 5 por ciento del PAAF eran citológicamente dudosos. La PAAF también proporciona un medio simple para establecer el diagnóstico de recidiva local o enfermedad metastática.
Diagnóstico
Histológico
La evaluación Histopatológica debe realizarse por un patólogo experimentado con un interés específico en la patología de los tejidos blandos. La distinción entre los sarcomas de alto grado (ejemplo, el fibrohistiocitoma maligno, el osteosarcoma extraesquelético) y las enfermedades no malignas (ejemplo, fascitis nodular y miositis osificante) puede ser sumamente difícil.
La reproducibilidad del diagnóstico histológico por un panel de expertos se evaluó en una revisión de 240 sarcomas. Después de la revisión, se reclasificaron histológicamente 61. El cambio predominante era de sarcoma, tipo no especificado, a un tipo específico. Se reclasificaron tres leiomiosarcomas a fibrohistiocitoma maligno y 4 de 10 fibrosarcomas se reclasificó como fibrohistiocitoma maligno. De importancia mayor, 12 de los 240 tumores (5 por ciento) habían sido clasificados como grados 3 o 4 fueron cambiados a grado 0 (lesiones benignas).
Resultados similares fueron anotados en un estudio del Grupo de Sarcoma de la Federación Francesa de Centros de Cáncer en el que se presentaron secciones normales de 25 sarcomas a un panel de 15 patólogos generales y un panel de patólogos que habían ayudado en el desarrollo del sistema graduación. La concordancia entre los grupos era sólo del 75 por ciento para el grado y 61 por ciento para el tipo histológico.
La evaluación del diagnóstico en casi todos los casos exige a una variedad de estudios inmunohistoquímicos para ayudar en la identificación del origen celular.
Inmunohistoquímica
de
los
tumores
de
partes
blandas
Los
tumores
de
partes
blandas
son
un
grupo
heterogéneo
de
tumores
con
una
apariencia
histológica
variable.
Siempre
existen
dificultades
en
el
diagnóstico
diferencial,
especialmente
en
el
caso
del
sarcoma
pleomórfico
de
alto
grado,
habiéndose
identificado
en
los
últimos
cinco
años
algunos
marcadores
inmunológicos
para
los
tumores
de
partes
blandas
y
preparado
anticuerpos
frente
a
ellos.
El
empleo
de
las
técnicas
de
inmunofluorescencia
sobre
tejidos
congelados
es
poco
práctica
debido
a
que
en
general
se
dispone
de
poco
tejido
y
a
que
el
detalle
morfológico
es
escaso.
Además,
muchas
zonas
antigénicas
se
destruyen
durante
los
procesos
de
fijación
e
inclusión
empleados
en
la
preparación
de
piezas
incluidas
en
parafina.
Para
la
detección
inmunohistoquímica
de
antígenos
en
tejidos
normales
y
en
tumores
se
emplean
con
mayor
frecuencia
técnicas
de
fijación
en
formalina
y
de
inclusión
en
parafina.
Tres
marcadores
son
útiles
para
mostrar
si
un
tumor
es
un
sarcoma,
un
carcinoma
o
un
linfoma;
de
entre
ellos,
el antígeno
leucocitario es
muy
específico
y
esta
restringido
a
la
superficie
de
las
células
de
la
serie
blanca,
siendo
sumamente
útil
para
distinguir
un
linfoma,
de
un
sarcoma
o
un
carcinoma.
Las citoqueratinas se
encuentran
en
tumores
de
origen
epitelial,
pero
pueden
encontrarse
también
en
algunos
sarcomas
como
el
sarcoma
epiteloide
y
el
sinovial.
Por
otro
lado,
el
filamento
intermedio
de vimentina se
encuentra
en
la
mayoría
de
los
tumores
mesenquimales
y
en
algunos
carcinomas.
Otros
antígenos
que
son
frecuentes
sólo
en
tumores
de
partes
blandas
son
la desmina (tumores
de
músculo
esquelético
y
liso), antígeno
asociado
al
factor
VIII (tumores
vasculares
benignos
y
malignos)
y
la proteína
S-100 (tumores
benignos
de
la
vaina
nerviosa,
tumores
de
células
granulares,
sarcomas
de
células
claras,
melanoma
y
algunos
tumores
neurales
malignos).
Algunos
de
estos
antígenos
también
se
encuentran
en
ciertos
tumores
primitivos
óseos.
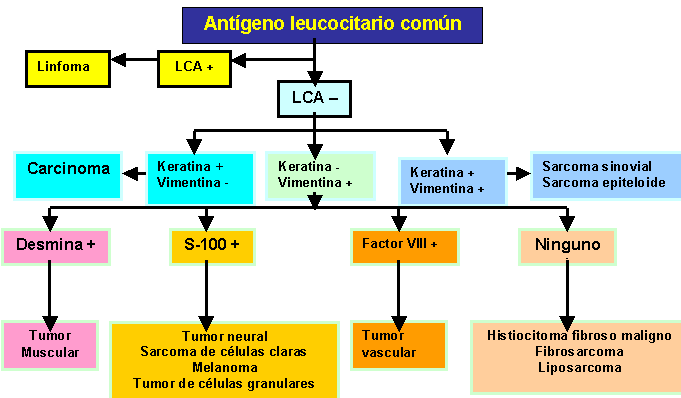{kind=link}
- El desmin es particularmente valioso en la identificación de células de los tumores de origen muscular: el rabdomiosarcoma y, en un grado menor, leiomiosarcoma.
- El antígeno S100 y los neurofilamentos hacen pensar en células que se originan de la vaina nerviosa.
- La citoqueratina puede ayudar distinguir entre sarcoma sinovial o sarcoma epitelial (qué contiene la citoqueratina) y fibrosarcoma (qué no tiene).
- El antígeno del factor VIII identifica tumores de origen endotelial.
Estadio
El
estadiaje
proporciona el
método
para
la
determinación
de
categorías pronosticas,
y
es
de
valor
enorme
en
la
interpretación
de
los
resultados
de
los
centros
de
tratamiento,
y
en
la
descripción
de
los
propios
resultados
de
un
centro.
La
decisión
acerca
de
la
estrategia
del
tratamiento
más
apropiada
se
influencia
fuertemente
por
el
estadio
en
el
diagnóstico
inicial.
Nosotros
recomendamos
el
sistema
organizado
para
sarcomas
de
tejidos
blandos
propuestas
por
el
American
Joint
Committee
on
Cancer (AJCC)
(ver tabla II).
El
sistema
de
estadiaje
de
la
AJCC
se
estructura
principalmente
en
el
grado.
La
importancia
del
grado
del
tumor
como
una
variable
del
pronostico
se
ha
demostrado
repetidamente;
la
supervivencia
disminuye
20
a
30
por
ciento
de
los
grados
1
y
2
a
los
grados
2
y
3.
El sistema de estadiaje de la AJCC incorpora los grados histológicos (G), tamaño del tumor (T), profundidad del tumor (superficial o profundo), metástasis ganglios linfáticos (N) y metástasis a distancia (M) para caracterizar cuatro agrupaciones de estadios. A cada tumor se le asigna una grado (1 a 2 o 3 a 4) en base a las características histopatológicas. El Estadio IV son definidos por cualquier afectación ganglionar regional o evidencia de enfermedad metastática a distancia. Si ambos están presentes, la lesión es la fase IV sin tener en cuenta el grado, tamaño o sitio.
Otra alternativa que del sistema de estadiaje se usa por algunas unidades quirúrgicas ortopédicas, incluyendo el sistema de estadios de la Musculoskeletal Tumor Society, y Memorial Sloan-Kettering (MSK). En el Surgical Staging System of the Musculoskeletal Tumor Society, el estadio es basado en el grado y compartimentalidad, esto es, el confinamiento a un compartimiento anatómico. Este parámetro es de valor en la predicción de la probabilidad del control local para el tratamiento por cirugía sola; sin embargo, no es un predictor exacto de recidiva local con la cirugía combinada con radioterapia. En una comparación de los sistemas de estadiaje para sarcomas de tejidos blandos localizados en las extremidades, los estadios de la AJCC o los de la MSK eran mas predictivos del riesgo de recaída sistémica.
Los
valores
pronósticos
de
un
sistema
de
estadiaje
para
sarcomas
principalmente
pertenece
a
su
valor
en
la
predicción
de
la
probabilidad
de
metástasis
a
distancia,
no
el
fracaso
local.
Para
este
ultimo
punto,
los
factores
importantes
son
el
grado
y
volumen
o
el
tamaño
del
tumor,
no
la
compartimentalidad.
En
una
revisión
de
factores
de
pronósticos
en
1041
pacientes
con
sarcomas
de
tejidos
blandos
en
las
extremidades,
el
tamaño
del
tumor
y
el
grado
eran
predictores
importantes
de
recurrencia
a
distancia
y
de
la
supervivencia,
mientras
la
edad
por
encima
de
50
años
y
la
presencia
de
márgenes
de
la
resección
positivos
predijeron
la
recidiva
local.
Además,
los
subtipos
histopatológicos
específicos
eran
predictivos
de
fracaso
local
(los
fibrosarcoma
y
Grado
Histológico
Se han desarrollado varios sistemas de graduación para aumentar el valor pronóstico de la valoración histológica. Para cada tipo histopatológico mayor, el pronóstico disminuye en pendiente con el grado (tabla III).
Tabla III: Tasa de metástasis a los 5 años en función del grado en 420 adultos con diferentes tipos histológicos de sarcomas de tejidos blandos.* |
|||
Tipo histológico |
Grado 1 |
Grado 2 |
Grado 3 |
Histiocitoma fibroso maligno |
(15) 100% |
(53) 75% |
(40) 49% |
Liposarcoma |
(22) 85% |
(24) 64% |
(11) 36% |
Leiomiosarcoma |
(12) 100% |
(18) 56% |
(13) 40% |
Schwanoma maligno |
(8) 100% |
(16) 60% |
(16) 34% |
Sarcoma sinovial |
(0) |
(22) 41% |
(17) 0% |
Sarcoma no clasificado |
(0) |
(13) 46% |
(41) 18% |
Otros |
(11) 100% |
(36) 43% |
(35) 39% |
| Los números entre paréntesis representan el número de pacientes en cada grupo. | |||
| Coindre, JM, Cancer Treat Res 1993; 67: 1 | |||
Los rasgos patológicos que determinan el grado incluyen celularidad, diferenciación, la abundancia de estroma, el pleomorfismo celular y nuclear, la presencia de necrosis, la atipia celular y nuclear, el número de mitosis y tipo histológico. La designación del grado es subjetiva, y dependiente a alguna magnitud en el patólogo específico. Hay normalmente poca discordancia en los sarcomas bien diferenciados o pobremente diferenciados. Los problemas aparecen con los tumores que están en la frontera entre grado 1 versus 2 y grado 2 versus 3. Hay celularidad creciente y pleomorfismo celular con los grados progresivamente más altos.
Tamaño del tumor
El tamaño del tumor se relaciona con la frecuencia de metástasis a
distancia así como el grado para, virtualmente, todos los tipos de tumores.
La
medida
normal
de
tamaño
del
tumor
es
el
diámetro
más
grande
de
la
lesión.
La
probabilidad
de
metástasis
distante
es
una
función
del
número
total
de
células
clonalmente
derivadas
y
el
volumen
del
tumor,
que
son
un
predictor
de
la
respuesta
específica
a
la
radioterapia. La importancia del tamaño en la frecuencia de
metástasis se ilustró en una revisión de 501 pacientes que lograron control
local de sarcomas de tejidos blandos. Para sarcomas grado 2 y 3, la
incidencia a cinco años de metástasis a distancia aumentó de 3 por ciento a
55 a 60 por ciento como el diámetro del tumor aumentado de 2.5 centímetro o
menos a >20 cm. Pocos
pacientes
desarrollaron
enfermedad
metastásica
en
sarcomas
de
grado
1.
Otros
Indicadores
Pronósticos
Aunque el grado histológico y tamaño del tumor son de valor pronóstico, hay una necesidad mayor de predecir el resultado para el paciente individual que es ahora factible. Entre los indicadores en estudio está el contenido de ADN celular, ploidía, cinética de la proliferación celular, y activación del gen, amplificación, supresión, o mutaciones.
Citometría de flujo del ADN - pueden usarse las citometrías de flujo para medir ADN contenido con manchas fluorescentes que se unen estoicométricamente al ADN. Un valor se establece para una población normal de células que proporcionan a un histograma gráfico las proporciones relativas de células 2N ADN ( esto es, células diploides en G0-G1), 4N ADN ( células tetraploides en G2-M) y ADN contenido entre 2N y 4N, llamado "fracción S-fase" (SPF). El índice proliferativo es la fracción de células que están en S, o G2 o M. Para medir el contenido de ADN por célula, la citometría de flujo también puede identificar las células con cantidades anormales de ADN, llamada aneuploidía. Varios estudios han sugerido que la presencia de aneuploidía en las células del sarcoma y un SPF aumentado tengan importancia pronóstica de la probabilidad de metástasis a distancia. En otra serie, sin embargo, la aneuploidía no fue un factor de significancia pronóstica en análisis multivariados. Parte de éstos resultados aparentemente contradictorios pueden ser incrementados por la sensibilidad de células aneuploides del tumor a los agentes quimioterapéuticos. La falta de valore predictivo independientes de la aneuploidía puede reflejar su relación al grado del tumor. Por ejemplo, en una serie, la frecuencia de aneuploidía aumentó de 8 por ciento con la el grado 2 a 89 por ciento con el grado 4.
Actividad proliferativa. Varias técnicas pueden evaluar la actividad proliferativa de un tumor que puede ser un indicador del potencial para las metástasis a distancia. Una de estas técnicas es la tinción de células por el anticuerpo monoclonal Ki-67 que reacciona con un antígeno nuclear presente en las células con el ciclo celular activo. Las células Ki-67 negativo no están en el ciclo celular activo (G0). El aumento del manchando nuclear por Ki-67 puede ser un marcador del pronostico independiente en los pacientes con sarcomas de los tejidos blandos. Un estudio, por ejemplo, evaluó el tejido de 121 pacientes con sarcoma de tejidos blandos de la extremidad de alto grado. La ausencia de metástasis a distancia a los 5 años en pacientes cuyos tumores tenían manchando nuclear con Ki-67 fue <20 comparados a >20 por ciento eran 70 y 50 por ciento, respectivamente. Otro indicador de actividad de proliferación celular que puede tener el valor pronostico es un anticuerpo monoclonal contra PCNA (proliferative cell nuclear antigen)/cyclin, una proteína nuclear S-fase-asociada. En una serie, la ausencia de metástasis a distancia en 3 años era mucho más alta en los pacientes con un PCNA bajo comparado a PCNA alto (87 contra 14 por ciento).
Oncogenes. Varios otros marcadores biológicos como la
súper-expresión
de
la proteína
p53,
ciertas
mutaciones
en
el
p53,
y
amplificación
del
oncogén
MDM2
que
ata
y
vuelve
inactivo
el
p53,
ha
sido
asociado
con
el
sarcoma
de
tejidos
blandos
de
grado
alto
y
la
supervivencia
pobre.
En
una
serie,
el
riesgo
relativo
de
muerte
era
notablemente
elevado
en
los
pacientes
con súper-expresión
p53
y
"
MDM2.
Sin
embargo,
estos
factores
no
se
han
mostrado
para
ser
indicadores
independientes
de
prognosis
todavía
y
no
se
han
usado
todavía
clínicamente
en
la
graduación
u
estadiaje
de
sarcomas
del
tejido
blando.
Diagnóstico molecular. Las SYT-SSX Fusión-Transcripciones. Actualmente se pueden confirmar e identificar sarcomas que se presentan en localizaciones raras así como en grupos de edad raros. Estamos en la era del diagnóstico molecular y no solo del diagnóstico histopatológico.
La mayoría de los sarcomas sinoviales celulares se caracterizan por la translocación t(x;18)(p11.2;q11.2) que funciona como un regulador de la transcripción aberrante. Los sarcomas sinoviales tienen 100% asociación con SSX1, SSX2 o SSX4 1A. El punto de ruptura de este translocación-fusión es el gen SYT del cromosoma 18 a uno de dos genes homólogos, SSX1 o SSX2 en el cromosoma X. Los sarcomas Sinoviales son el 10 por ciento de los sarcomas de tejidos blandos e incluyen dos subtipos histológicos mayores, bifásico y monofásico, definidos respectivamente por la presencia o ausencia de diferenciación de epitelio glandular. En un estudio de 45 sarcomas sinoviales analizados por reacción en cadena con polimerasa inversa, había una correlación significativa entre el tipo patológico (monofásico o bifásico) y SSX1 o SSX2 en la transcripción de fusión. Todos los tumores bifásicos tenían las SYT-SSX1 fusión transcripción, y todos los 16 tumores que eran SYT-SSX2 eran monofásicos. La supervivencia metástasis-libre era significativamente más alta en los 24 pacientes con la SYT-SSX1 fusión transcripción en el análisis multivariado de los 39 pacientes sin metástasis. Microchip de DNA se están empleando ahora en los estudios de diagnóstico del sarcoma.
El
gen
de
la
familia
del
sarcoma
de
Ewing
(EWS)
se
ha
identificado
en
el
desarrollo
de
sarcomas.
También
juega
un
papel
en
el
desarrollo
de
PNET
(tumor neuroectodérmico
periférico
que
nosotros
sabemos
ahora
que
es
idéntico
al
sarcoma
de
Ewing)
y
al
sarcoma
de
células
claras.
Como
el
diagnóstico
se
hace
en
un
nivel
molecular,
se
han
catalogado
los
diferentes
tumores,
combinando
algunos
y
excluyendo
a
otros.
Sabemos
ahora
que
el
condrosarcoma
mixoide
del
esqueleto
no
es
la
misma
enfermedad
que
el
condrosarcoma
mixoide
extraesquelético
Discusión
El
enfoque
del
tratamiento
de
los
tumores
de
tejidos
blandos
es
multidisciplinario.
Como
dijo
el Dr
Antonio
Herrera
Rodríguez:
“si
no
disponemos
de
los
medios
necesarios
para
tratar
los
tumores
del
Aparato
Locomotor,
veámoslos
pero
no
los
toquemos".
Debido a que la mayoría de los sarcomas de partes blandas son de crecimiento lento y generalmente asintomáticos, pueden ser infravalorados, practicándoles biopsias excisionales con márgenes inadecuados que alteran significativamente el tratamiento y el pronóstico futuros.
El tratamiento adecuado de un paciente con un sarcoma de partes blandas comienza con una buena historia clínica y exploración física, así como un estudio de imagen adecuado para valorar el estadio de extensión local y a distancia. La biopsia se realizará por un cirujano experimentado y no debe interferir en la cirugía definitiva. Es obligatorio que el paciente sea tratado por un equipo multidisciplinario en un centro de referencia.
El
tratamiento
y
pronóstico
de
los
sarcomas
de
partes
blandas
depende
principalmente
del
estadio
del
mismo
más
que
del
diagnóstico
histológico.
Todos
los
sistemas
de
estadificación
tienen
en
cuenta
el
grado
de
diferenciación
del
tumor
(término
histológico
que
se
relaciona
con
el
grado
de
diferenciación
del
tejido
tumoral
en
comparación
a
un
tejido
embrionario
o
adulto
conocido
y
relacionado
con
el
mismo).
Cirugía
La cirugía como primer paso en el tratamiento de los tumores de partes blandas tiene como ventaja:
1. una mejor cicatrización por primera intención
2. la posibilidad de disponer de todo el espécimen para el análisis anatomopatológico
3. identificar
y
marcar
sitios
de
dudosa
resección
con
clips
radioopacos.
La
cirugía
conservadora
combinada
con
la
radioterapia
y/o
quimioterapia
consigue
resultados
similares
a
la
cirugía
más
radical.
OBJETIVOS
1. Obtención del control local del tumor. La recidiva local condiciona una cirugía posterior más agresiva y una incidencia mayor de metástasis.
2. Obtención de un resultado funcional bueno. La cirugía radical se reserva para lesiones extracompartimentales o para tumores intracompartimentales grandes. La resección local debe ser amplia, sin ver la superficie del tumor. El espécimen resecado debería estar rodeado completamente de una envoltura musculofascial intacta. Los músculos deben ser seccionados cerca de sus orígenes de inserción para eliminar áreas de potencial extensión longitudinal del tumor. Si hay que resecar hueso, nervios o arterias de gran calibre, se debe hacer, si es posible, una resección en los planos subperiósticos, subepineural o subadventicial. Si a pesar de todo los márgenes son positivos y se recurre a la amputación, esta debe hacerse proximal al compartimento comprometido, con el fin de evitar la recurrencia en el muñón. La amputación es más eficaz para el control local del tumor pero no previene el riesgo de metástasis. La radioterapia mejora de forma substancial el control local de los tumores de partes blandas cuando se dejan fragmentos irresecables, por eso la mayoría de los autores no recomiendan la amputación y esta solo se reserva para casos de recidivas no resecables después del tratamiento combinado con cirugía y radioterapia.
Para los pacientes tratados en otro centro, hay autores que propugnan la realización de una nueva cirugía para comprobar los márgenes con biopsias intraoperatorias e instaurar tratamiento con radioterapia intraoperatoria si es necesario.
En
algunos
centros
hay
la
posibilidad
de
irradiación
intersticial
mediante
la
colocación
de
tubos
finos
en
el
lecho
del
tumor,
que
permiten
la
irradiación
posterior
con
iridio192.
Radioterapia
El papel de la Radioterapia en el tratamiento de los tumores de partes blandas es muy importante. Puede ser:
1. Postoperatoria, fue la primera en aplicarse. Se utiliza después de la resección local para acabar con los restos microscópicos del tumor. En la actualidad la radioterapia postoperatoria está indicada para los pequeños tumores en los que solamente pueden lograrse márgenes adecuados mediante la amputación.
2. Intraoperatoria.
Ventajas:
- Una buena limitación del área de enfermedad residual
- Permite la radioterapia y resección quirúrgica simultaneas
- Menor toxicidad ya que se pueden proteger las estructuras sensibles
- Disminuye el campo a irradiar en el postoperatorio.
- Durante el acto quirúrgico se puede implantar catéteres para braquiterapia, permitiendo una radiación adicional en el lecho tumoral. Alternativamente también se puede utilizar radioterapia intraoperatoria externa convencional
Desventajas:
- Su mayor inconveniente es la neurotoxicidad. En la casuística expuesta la mayoría de los pacientes que desarrollan complicaciones recibieron dosis altas (2000 cGy), por lo que las dosis óptimas están en el rango de 1000 a 1500 cGy
3. Preoperatoria. En la última década se ha empezado a utilizar la radioterapia preoperatoria sola o combinada con adriamicina intraarterial. La radiación preoperatoria se administra generalmente después de la biopsia incisional y se administran dosis totales de 5000 a 5200 cGy.
Ventajas:
- Inactivación de gran cantidad de células tumorales, y con ello disminución de la diseminación intraoperatoria.
- Debilitación de las células que entran en el torrente sanguíneo. Con ello se disminuye su capacidad de producir metástasis a distancia.
- En los tumores de gran tamaño se consigue la reducción del tamaño, y la necrosis periférica permitirá una resección mas fácil (durante la resección se elimina menor cantidad de tejido sano) sin riesgo de diseminación local.
- Disminuye el campo a irradiar en el postoperatorio.
- Con este abordaje la tasa de recurrencias de los sarcomas de partes blandas es del 6%
- Cuando el sarcoma es de gran tamaño y está localizado junto a un paquete neurovascular o al hueso está indicada la radioterapia preoperatoria con o sin quimioterapia intraarterial.
Desventajas
- La tasa de problemas de cicatrización de las heridas es del 20%. Estas consideraciones son importantes en los tumores de gran tamaño adyacentes a estructuras neurovasculares o al hueso.
La
cirugía
definitiva
se
realiza
3
semanas
después
de
completar
la
radioterapia.
Después
de
que
la
herida
ha
cerrado
se
administra
tratamiento
adicional
al
lecho
tumoral
hasta
completar
una
dosis
total
de
6400
a
6600
cGy.
Si
se
utiliza,
además
,
adriamicina
intraarterial
la
dosis
total
de
radiación
puede
ser
disminuida
y
la
dosis
darse
a
intervalos
menores.
La
dehiscencia
de
la
herida
y
la
intensa
fibrosis
de
la
extremidad
han
sido
los
mayores
problemas
con
este
tipo
de
terapias.
Quimioterapia
La
introducción
de
la quimioterapia ha
mejorado
notablemente
el
pronóstico
al
reducir
el
riesgo
de
recidiva
local
y
de
metástasis
a
distancia.9,12,39,41,46cot40 La
pauta
que
se
está
extendiendo
es
la
de
practicar
quimioterapia
neoadyuvante,
cirugía
de
márgenes
amplios
o
radicales
y
quimioterapia
adyuvante.
Actualmente
existen
tres
grandes
tipos
de
terapia
preoperatoria:
la
quimioterapia
intravenosa,
la
quimioterapia
intraarterial
y
la
radioterapia.
1. La quimioterapia intravenosa es la que tiene los mejores efectos sistémicos sobre las micrometástasis, mientras que la radioterapia local no tiene efectos sistémicos y actúa únicamente sobre el tumor primario.
Ventajas del uso de la quimioterapia preoperatoria intravenosa en pacientes con sarcomas.
- Posibilidad de iniciar un tratamiento sistémico sin demora.
- La quimioterapia preoperatoria y la radioterapia posibilitan una regresión del tumor primario que facilitará la conservación de la extremidad. Además la vascularización perineoplásica y el edema generalmente se resuelven con una terapia efectiva permitiendo al radiólogo realizar una nueva imagen del tumor y definir mejor la extensión de la masa.
- La terapia preoperatoria, en algunos tumores, hace que sea posible medir la efectividad de algún tipo de terapia contra el tumor primario o metastásico in vivo. La quimioterapia sistémica se justifica por la probable existencia de micrometástasis no detectables clínicamente en el momento del diagnóstico. El fármaco más efectivo en el tratamiento de los sarcomas de partes blandas es la adriamicina
- La quimioterapia intraarterial puede ser muy efectiva para el tumor local, pero su efecto sistémico en las micrometástasis es menor que la quimioterapia intravenosa.
- La quimioterapia postoperatoria esta, en el momento actual considerada, como experimental o para el uso de casos individuales
Los paciente con enfermedad diseminada (estadio III) tienen un pronóstico malo ya que no se dispone de ningún protocolo curativo.